随着长读长、全长 RNA 测序方法的不断开发,科学界已逐渐认识到一种基本范式转变——由于频繁生成来自单个基因组位点的转录本,对人类转录组的理解方式从“以基因为中心”转变为“以异构体为中心”(Jing et al. (2019) Oncogene 38: 3047)。 PacBio 推出的 Iso-Seq 方法已在众多论文中得以应用,让研究人员对转录组的复杂性及其对理解生物学和疾病的影响有了一些新的见解。 与短读长 RNA-seq 仅可观察转录本的小片段不同,长读长、全长 RNA 测序能够直接观察整个剪切异构体。
最近,Weill Cornell Medicine 旗下的 Hagen Tilgner 实验室及其合作者在一篇新论文中描述了对用于全长 RNA 测序的各种长读长技术的详细对比,他们采用了一种非常巧妙的条形码策略,通过不同的测序技术对来自相同 RNA 分子的 cDNA 拷贝进行了测序。
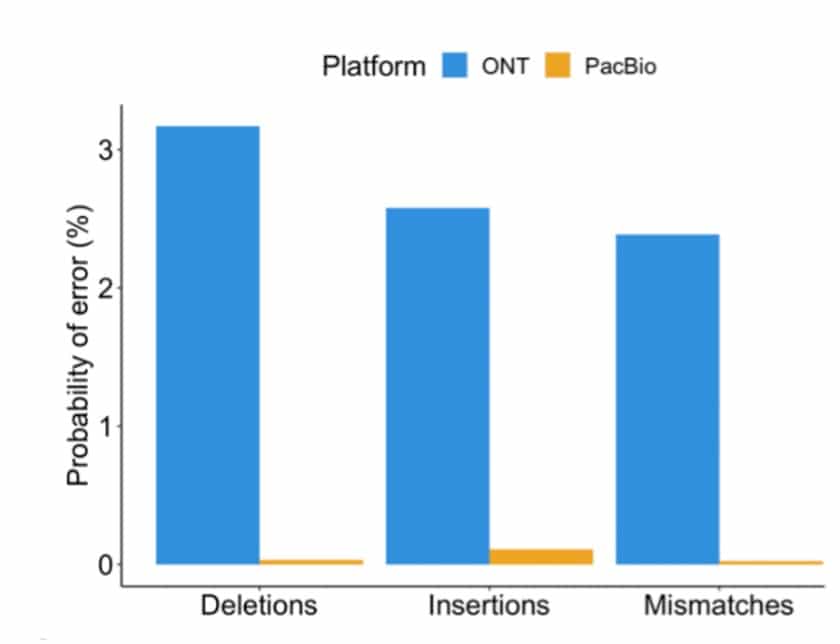
如这篇论文所述,PacBio HiFi Iso-Seq reads 明显更为精准(见下图 准确率较低的 ONT reads 会导致序列错误诱发的不准确 ONT 比对,包括短外显子比对缺失。 PacBio 的高 read 准确度也带来了更高的条码回收率,PacBio 的回收率可达 85%,相比之下, ONT 的回收率仅为 16%。 显然,大多数 PacBio 转录本 reads 也比 ONT 长,而且通常包含对定义完整异构体而言十分重要的元素。 作者还发现,PacBio reads 在识别转录本起始位点、剪切点和短外显子定位方面表现优异。
关于短读长 RNA-seq,在 OHSU 研究人员发布的一篇新预印本论文中,使用了 PacBio 的 Iso-Seq 数据作为真实基线,评估了从短读长数据中检出内含子保留转录事件的能力。
结果表明,短读长 RNA-seq 的事件检出能力略有不足。作者总结道:“我们发现,用短读长工具检测内含子保留的查全率低,查准率更差,这让我们对使用常用方法检出的大部分推定保留内含子的完整性和有效性产生了怀疑。”
PacBio 的 Iso-Seq 方法保障了 cDNA 测序的最高质量,增强了我们探索有关转录组的组成、动力学和疾病相关畸变的基础数据的信心。 在近期的一篇论文中,Jacques Banchereau 博士(杰克逊实验室)及其合作者为我们展示了如何应用 PacBio 的 Iso-Seq 方法为乳腺癌创建综合转录本异构体资源,并发现了数千起乳腺肿瘤特异性剪切事件,其中 35 起事件与患者存活率显著相关(其中 21 起不在 GENCODE 中,10 起在特定的乳腺癌亚型中很常见)。
