
揭开复杂群落和微生物组的秘密是微生物学中最令人兴奋的前沿领域之一,有可能彻底改变我们对人类健康、食品生产和环境保护的认识。为了给这种突破奠定基础,充分捕捉和分析微生物组或复杂群落中存在的所有遗传信息至关重要;而鸟枪法宏基因组测序法对于获得这种洞察力至关重要。
为了追求最好的鸟枪法宏基因组数据,PacBio HiFi测序继续为质量和完整性设定标准,这要归功于数据分析工具中令人兴奋的创新。这些新的管道,与HiFi数据的高精确度和长读数长度的有效结合,产生了比市场上任何其他技术更精确的分析信息。此外,HiFi数据比以往任何时候都能返回更多的循环、单片段元基因组组装基因组(MAGs),让你有能力发现隐藏的微生物生态系统功能,并自信地捕捉物种的丰富性和多样性。
- HiFi鸟枪法宏基因组测序产生的数据集由单个读数组成,最多可跨越8个连续的基因,使您能够一次性精确地剖析一个群体的功能和分类学。
- 得益于下采样实验的启示,HiFi宏基因组的分类学和功能分析现在可以在不影响准确性的情况下以近1/3的成本†进行。
- 以组装为重点的HiFi宏基因组研究可以得到几十到几百个高质量(HQ)的MAGs,其中高达33%的MAGs为单株,使您可以预测基因并进行权威的分类学注释。
以较低的成本获得高质量的HiFi宏基因组分析数据
剖面图更具有成本效益,只用0.5Gb的高保真数据就能获得与88Gb,或可能更多的最先进的物种和多样性信息。
研究证明:为了帮助微生物学家从HiFi数据中获得最具成本效益的见解,我们进行了一系列下采样研究。每次分析都是为了找到实现非常准确的群落剖面图所需的最小深度,并确定HQ和单重叠MAG恢复率。
首先,我们利用TruMatrix技术对ZymoBIOMICS粪便参考物进行了测序,这是一个高度复杂的人类肠道微生物样本集合,对于基准测试和参考标准非常有用。我们使用4个SMRT Cell 8M对该样本进行测序,然后对数据集进行逐步降采样,使其覆盖范围相当于在单个SMRT Cell8M(96丛)上运行96个多路测序。该研究总共捕获了88到0.3 Gb的数据。然后通过PacBio的分类分析工作流运行每个数据级别。
首先,我们用TruMatrix技术对ZymoBIOMICS的粪便参考样本进行了测序,这是一个汇集的高度复杂的人类肠道微生物组样本,对基准和参考标准非常有用。我们用4个SMRT Cell 8M对该样本进行了测序,然后将数据集逐步下采样,使其覆盖率与单个SMRT Cell 8M(96-plex)上的96次复用测序运行相当。该研究采集了88至0.3千兆字节(Gb)的数据。然后通过PacBio的分类分析工作流程运行每个数据级别。
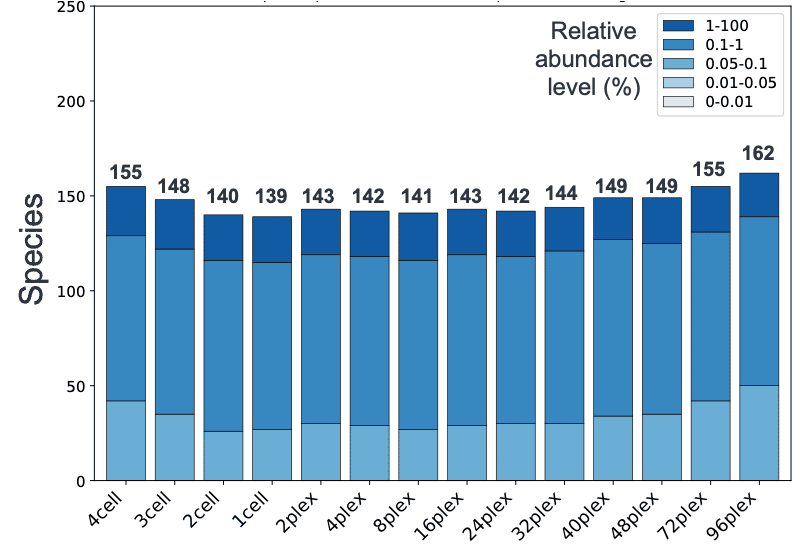
该工作流程的结果表明,从4个SMRT细胞8Ms到48个丛获得的信息基本一致,不仅恢复的物种数量相似(图1),而且相对丰度分布也几乎相同(图2)。这表明,0.5 Gb的48倍级别上可以获得与88 Gb相同级别的分类学特征信息,为微生物组研究人员提供了进入HiFi宏基因组学世界的更实惠的切入点。
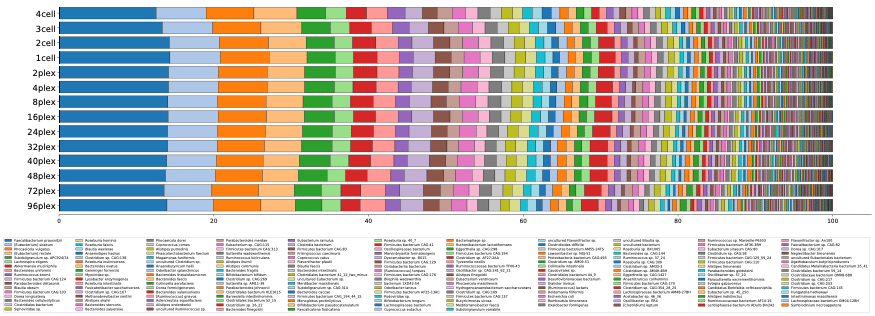
用比以往任何时候都多的HQ MAG捕捉物种的精细细节
从每个SMRT Cell 8M的一个样本开始,实现以装配为中心的宏基因组HiFi测序的最佳成本效益。寻求更深入了解的项目可以增加多个SMRT Cell 8M的测序深度,以生动详细地捕获稀有微生物。
我们如何获取:我们下采样研究的后半部分致力于建立以宏基因组组装基因组(MAG)为重点的研究指南。为了评估测序深度对MAG组装的影响,我们采用了ZymoBIOMICS TruMatrix 4 SMRT Cell 8M肠道微生物组数据集,并逐步将其降采样至8倍深度,以查看各个水平对组装和恢复的影响。我们知道组装对测序深度更为敏感,测序深度和结果之间存在非常可预测的关系。然而,在我们的管道中,深度对HQ-MAG回收率和单个连续MAG回收率的影响是不同的。
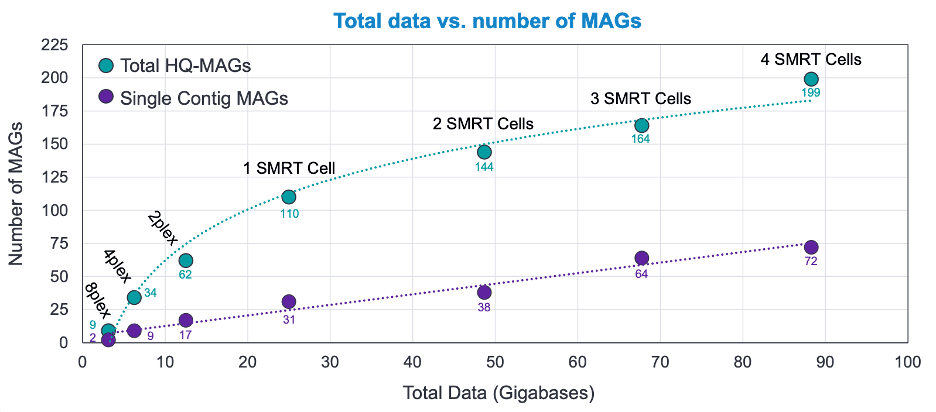
我们工作流程的结果显示,从8-plex开始,MAG的总回收量和深度之间有一个对数关系(图3中的绿线),迅速增加到一个SMRT Cell 8M。从这里开始,随着测序深度的增加,回收的效率有所下降,但HQ-MAGs仍然大量返回,最深的测序产生的几乎可以肯定是更罕见的物种。对于单片段MAG的回收(图3,紫线),有一个明显的回收与深度的线性关系,4个SMRT细胞8Ms共产生199个HQ-MAG,其中72个是单片段。即使在8-plex的深度,也有9个HQ-MAGs被回收,其中2个是单一的contig。在4-plex中,产生了34个HQ-MAGs,其中9个是单株。这些结果清楚地表明,HiFi鸟枪法宏基因组测序可以提供大量的和惊人的详细的基因组组合,这在其他测序技术中是难以获得的。
极其清晰地窥视微生物世界
HiFi宏基因组学使微生物组研究人员有能力以比以往更高的成本效益精确地描述一个社区的功能和分类。同时,HiFi reads令人难以置信的长度和准确性使研究人员能够利用令人印象深刻的HQ-MAG恢复来预测基因并以更大的信心进行分类注释。这些优势加在一起,使HiFi测序成为旨在实现突破性发现的雄心勃勃的项目的首要测序解决方案。
