新优化的生物信息学流程支持科学家利用 HiFi 测序技术检测比以往更多的物种,组装更多的单 contig 宏基因组——某些案例中的增幅可达 9 倍,这对微生物和宏基因组学爱好者而言无疑是个令人振奋的消息。
得益于 PacBio HiFi 长读长测序技术的不断进步,结果质量的提升以及成本效益,它日益成为复杂微生物群落宏基因组分析的流行工具。
最近,PacBio 团队专注于更新工具和改进流程,以确保这两方面针对 HiFi 数据完全优化,为宏基因组研究提供更多价值。
努力终究会带来回报, 经过团队人员的不懈努力,宏基因组学分类分析现能够以更高精确度获得更多分类的低丰度物种,鸟枪法宏基因组组装可产生更多高质量、完整的环状单 contig 宏基因组组装的基因组 (MAG)。
挖掘数据
为测试新流程的效力,我们使用来自 BioCollective 的混合人肠道微生物组样本进行了一项研究。
其中,2 份样本来自素食供体,2 份样本来自杂食供体,以支持我们了解膳食如何影响肠道微生物。 通过合并多个供体(在此案例中为 4 名成年人)的样本来创建参考材料的合并过程,由此产生的样本要比模拟群落方法更为复杂,且同时拥有更为丰富的数据集。 相较于单个供体来源的样本,通过该方法产生的样本组成也更加一致。 该数据集可供任何人使用。
每份样本均通过 Sequel II 系统在一个 SMRT Cell 8M 上完成测序,每份样本大约产生 200 万条 reads,平均读长接近 10 kb,中位数质量值为 Q40。
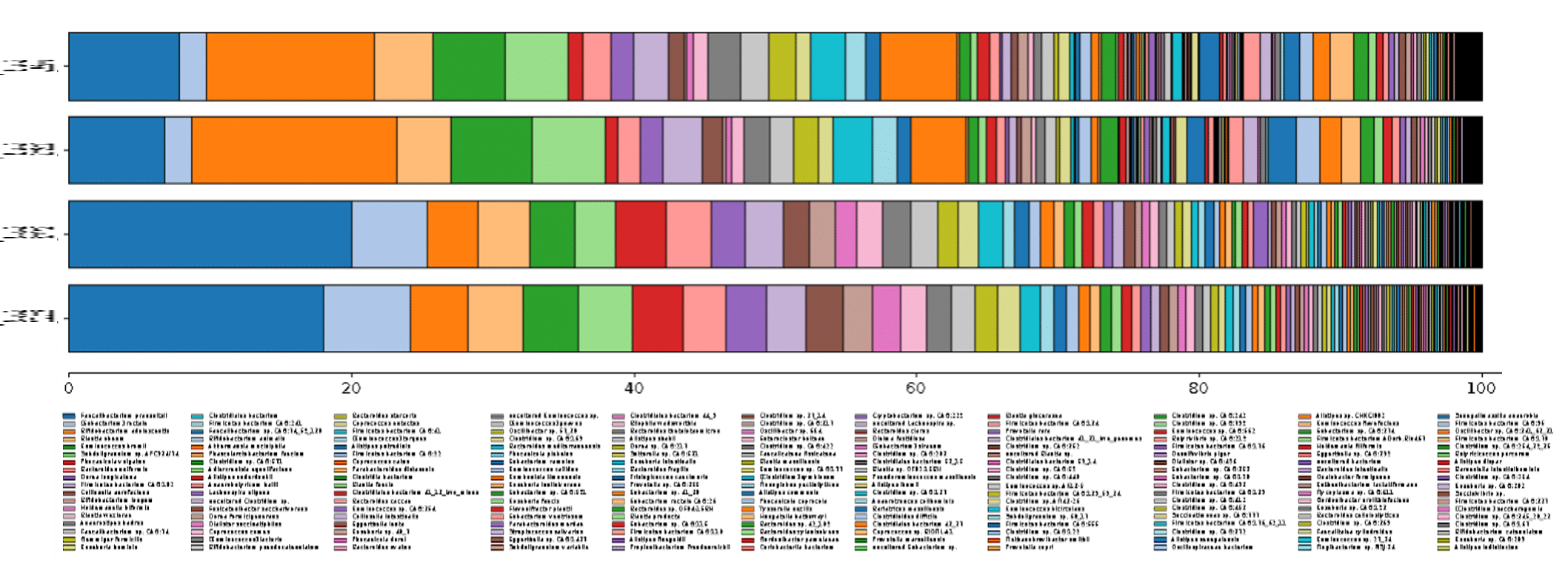
利用更新的生物信息学流程,我们进行了鸟枪法宏基因组分类分析,在优化的检测设置检测到的物种数量可达 199 个,在超灵敏设置下检测到的物种数量可达 690 个(图 1)。
此前的工作在保守检测设置下仅检测到了 76 个物种,与之相比,从同一样本分类中检出的物种增加了 123-614 个,按百分比换算可达 162%-808%!
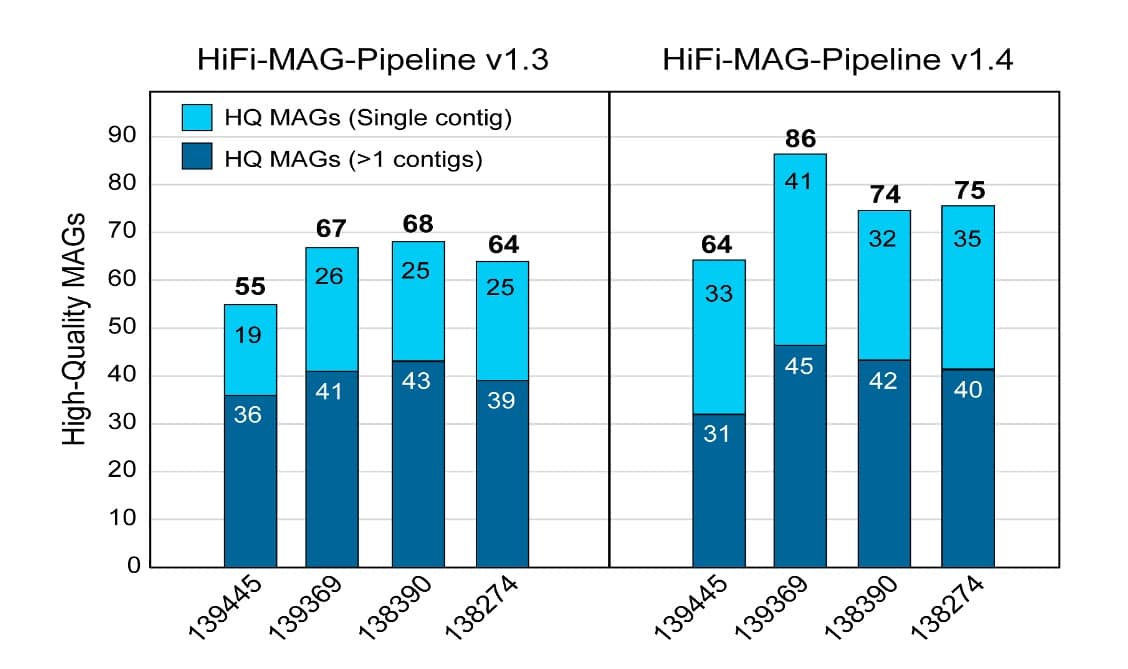
随后,我们进行了鸟枪法宏基因组组装,目标是获得至少 70% 完整的高质量 contig MAG,污染少于 10%,低于 10 个 contig。 虽然在此前的研究中,每份样本产生了 55-70 个高质量 MAG,其中 25 个包含在单个 contig 中,约 126 个物种/菌株,但使用更新后的生物信息学流程结合新型环状分箱策略共产生了每份样本 65-85 个高质量 MAG,其中单 contig 数量达 35 个,约 143 个物种/菌株(图 2)。
也就是说,来自同一数据的高质量 MAG 增加了 45 个,单 contig MAG 增加了 46 个,分别增加了 18% 和 48%。
这些改进将帮助我们的客户从样本数据以及从每个 SMRT Cell 中获得更多信息,这令我们十分欣慰。 新技术带来的真正高质量的 MAG 以及无需组装的单 contig 基因组可提供更多见解,加速科学发现,这一切都值得庆祝。
如需进一步了解如何将 HiFi 整合到宏基因组的工作流程,请访问我们专门的微生物学 和复杂群体页面,以及 HiFi 测序概述。
