
最近,我们邀请 PacBio 微生物组和宏基因组学市场总监 Jeremy Wilkinson 博士 、Dana-Farber 肿瘤研究所及 Harvard Medical School 博士后 Xiaowen Feng 博士 以及 PacBio 资深生物信息学科学家 Daniel Portik 博士 共同探讨了 PacBio HiFi 宏基因组学数据处理流程的最新进展。
从这场网络研讨会能够看到,结合 PacBio HiFi 测序,这套新的分析工具所能提供的数据具有无与伦比的质量和宽度,助力微生物学家 获得前所未有的微生物群落的分类细节和功能信息水平。
重点信息:
-
新开发的 hifiasm-meta 软件工具支持 PacBio HiFi 宏基因组学流程正确识别 比其他现有流程更多的环状单 contig MAG 和总 MAG。
- 借助 PacBio HiFi 测序 reads,宏基因组学研究就能 获得物种水平甚至株系水平的分类分辨率以及功能信息。
- HiFi 宏基因组学下采样实验表明,一份样本在 4 个 SMRT Cell 上运行和 48 份多重分析的样本在一个 SMRT Cell 上运行,具有几乎相同的数据宽度,支持 最大化成本效益,同时也能从样本中获得最新见解。
“HiFi reads 能让我们产生几乎完整的宏基因组图景,而不只是片段化的组装”-Bickhart et al.
Hifiasm-meta 增强 HiFi 宏基因组学的能力
在研讨会开始时,Xiaowen Feng 介绍了关于鸟枪法宏基因组学的最新进展。 Xiaowen Feng 谈到,宏基因组样本的 <102>De novo</102> 组装是研究微生物群落的常用方法。 同时,她指出了短读长组装的明显局限性,以及它们在分析过程中的分箱如何推动了 metaFlye 的开发,metaFlye 是唯一一款已发布的专门用于长读长宏基因组组装的组装程序。 但是,为了充分利用 HiFi reads 长而准确的优势,Xiaowen Feng 和同事们开发了hifiasm-meta,将更早期工作扩展到宏基因组学样本。 对 7 个经验数据集的评估结果表明,hifiasm-meta 重建了每个数据集中数十个到数百个完整环状细菌基因组,其中一些基因组 >1 Mb,性能始终优于其他宏基因组组装程序。
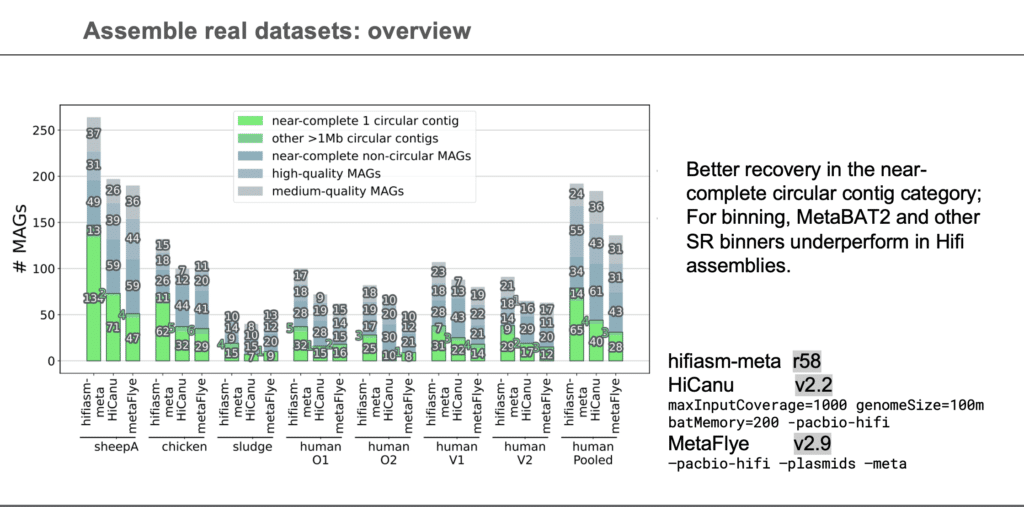
HiFi 宏基因组学能以极高的精准度同时获得宏基因组的分类和功能信息。
Daniel Portik 使用来自 BioCollective 的 4 份混合人肠道微生物组样本 演示了最新优化的 PacBio 生物信息学 流程的能力。 利用专为 HiFi reads 定制的分析,进行了分类和功能分析以及宏基因组组装。 以优化高查全率和查准率为目标,进行分类分析设置,在 4 份样本中共检出 199 个物种。 而放宽分析设置后,总计检测到多达 690 个物种。 使用 Hifiasm-meta 与 PacBio 分箱流程联合进行宏基因组组装,以鉴定并表征高质量 (HQ) 宏基因组组装的基因组 (MAG)。 Hifiasm-meta 支持的工作流程在 4 份样本中总共鉴定了 299 个 HQ MAG(>70% 完整,<10% 污染,<20 个 contig)。 其中,141 个 MAG 包含单个环状 contig。 最后,对数据进行了向下采样,多种多重分析方式,并研究了对这些分析的影响。 无论是 4 个 SMRT Cell 还是 48 份多重分析样本在 1 个 SMRT Cell 上运行,物种检测和功能分析结果都十分可靠。
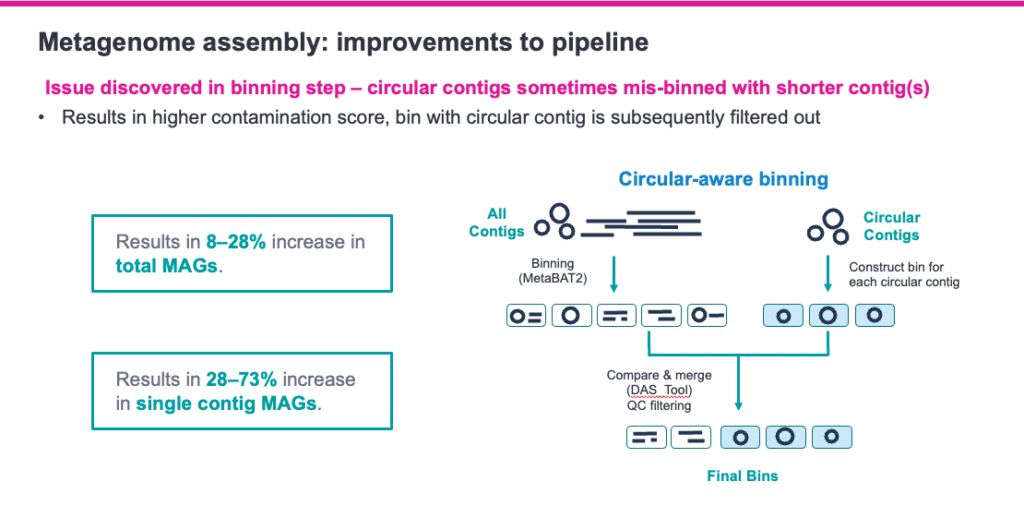
PacBio HiFi-MAG-Pipeline 的环状分箱特性支持从样本处获得更多 MAG 和单 contig MAG。
问答环节精选内容
问题: 使用 HiFi reads 进行 MAG 分箱是否有“合适的”程序?
回答: 根据测试,MetaBAT2 似乎是目前为止最好的 HiFi reads 分箱工具,但我们建议尝试所有分箱程序,看哪个程序能对您的数据产生最佳结果。
问题: 对于基于 reads 的分析,与短 reads 结果进行过比较吗?
回答:
最近有 2 篇论文做了这种比较,分别为 Gehrig et al 2022 和 Portik et al 2022。在检测到的物种数量方面,二者在过滤后差不多,结果相似,物种一致。 在查准率和查全率方面 PacBio reads 优于短 reads,这是因为 kmer 方法精准度不足,即使在过滤后,也无法达到 HiFi reads 从上述流程中直接获得的相同水平的查准率和查全率。 更多的 PacBio 读段被分配给功能,通常约 80%-90% 的 reads 被注释,而短 reads 有约 1/3 被分配。
问题: 大部分结果侧重于原核群落。 对于包含真核生物的宏基因组学群落,您预计 HiFi reads 的表现会如何?
回答: Priest et al 2021 是一个很好的案例研究,演示了此类环境。
问题: 对于宏基因组组装,是采用来自更丰富的生物体的闭环基因组,还是也从低丰度生物体获得单 contig MAG?
回答: 二者都有。 对于丰度更高的生物体,更容易获得闭环的基因组,但也可以对低至 5-10 倍基因组覆盖度的低丰度生物体获得闭环,在分析的多个数据集中,两种情况都有出现。
用 Pacbio HiFi 鸟枪法宏基因组学获得更好的结果
HiFi 宏基因组学为研究微生物群落的微生物学家提供了全新水平的数据质量、全面性和见解。 得益于最新优化的由 hifiasm-meta 支持的数据处理工作流程,研究人员可以正确鉴定<263>比任何其他现有流程更多的环状单 contig MAG 和总 MAG。 PacBio HiFi 测序 reads 可 获得物种水平甚至株系水平的分类分辨率以及功能信息,同时最大化成本效益。
